
Cell Signaling + Central dogma
Ligand - Receptor - Cascade pathway - Transcription (Pre mRNA, tRNA & rRNA) - Post Transcriptional Modification (Mature mRNA) - Translation (Amino acid Sequence) - Post Translational Modification - Protein Production - Cellular functions.
Prokaryotic Transcription
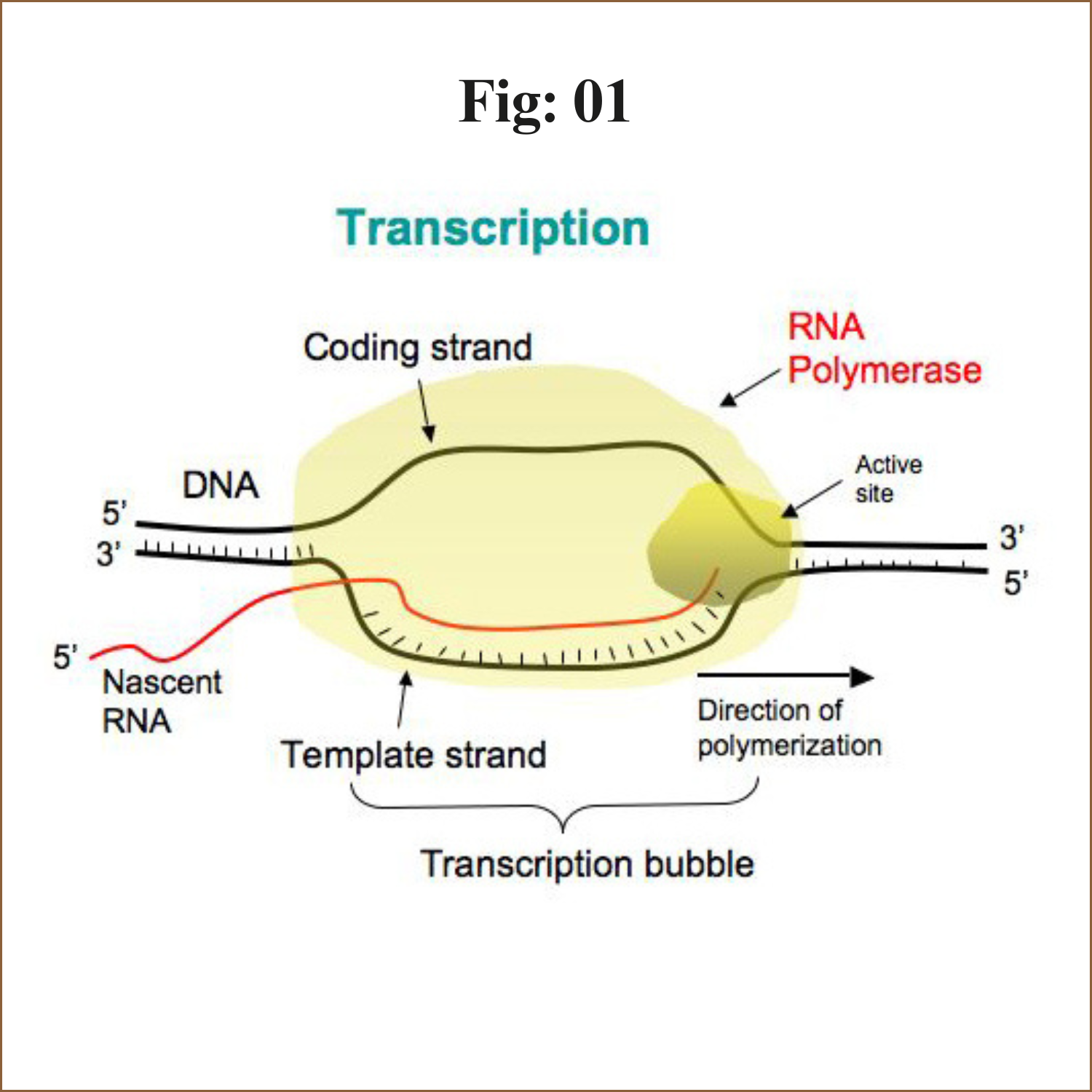
The process of synthesis of RNA by copying the template strand of DNA is called transcription.
During replication the entire genome is copied but in transcription only the selected portion of the genome is copied.
The enzyme involved in transcription is RNA polymerase. Unlike DNA polymerase it can initiate transcription by itself, it does not require primase. More exactly it is a DNA dependent RNA polymerase.
The steps of transcription
Transcription is an enzymatic process. the mechanism of transcription completes in three major steps
1. Initiation:
a) Closed complex formation
b) Open complex formation
c) Tertiary complex formation
2. Elongation
3. Termination:
a) Rho- dependent
b) Rho-independent
1. Initiation:
The transcription is initiated by RNA polymerase holoenzyme from a specific point called promotor sequence.
Bacterial RNA polymerase is the principle enzyme involved in transcription.
Single RNA polymerase is found in a bacteria which is called core polymerase and it consists of α, β, β’ and ω sub units.
The core enzyme bind to specific sequence on template DNA strand called promotor. The binding of core polymerase to promotor is facilitates and specified by sigma (σ) factor. (σ70 in case of E. coli).

The core polymerase along with σ-factor is called Holo-enzyme ie. RNA polymerase holoenzyme.
In case of e. coli, promotor consists of two conserved sequences 5’-TTGACA-3’ at -35 element and 5’-TATAAT-3’ at -10 element. These sequence are upstream to the site from which transcription begins. Binding of holoenzyme to two conserved sequence of promotor form close complex.
In some bacteria, the altered promotor may exist which contain UP-element and some may contain extended -10 elements rather than -35 elements.

Region1: it includes 1.2 and 1.1 region. Region 1.1 acts as molecular mimic of DNA
Region2: it recognizes -10 element in promotor. α-helix recognizes -10 element.
Region 3: it recognizes extended -10 element.
Region 4: it recognizes -35 element in promotor by a structure called helix-turn-helix.
The UP-element is recognized by a carboxyl terminal domain of α-sub unit called αCTD (carboxyl terminal domain) which is connected to αNTD (Amino terminal domain) by flexible linker.
Closed complex: Binding of RNA polymerase holoenzyme to the promotor sequence form closed comolex
Open complex: After formation of closed complex, the RNA polymerase holoenzyme separates 10-14 bases exztending from -11 to +3 called melting. So that open complex is formed. This changing from closed complex to open complex is called isomerization.
Tertiary complex: RNA polymerase starts synthesizing nucleotide. It doesnot require the help of primase.
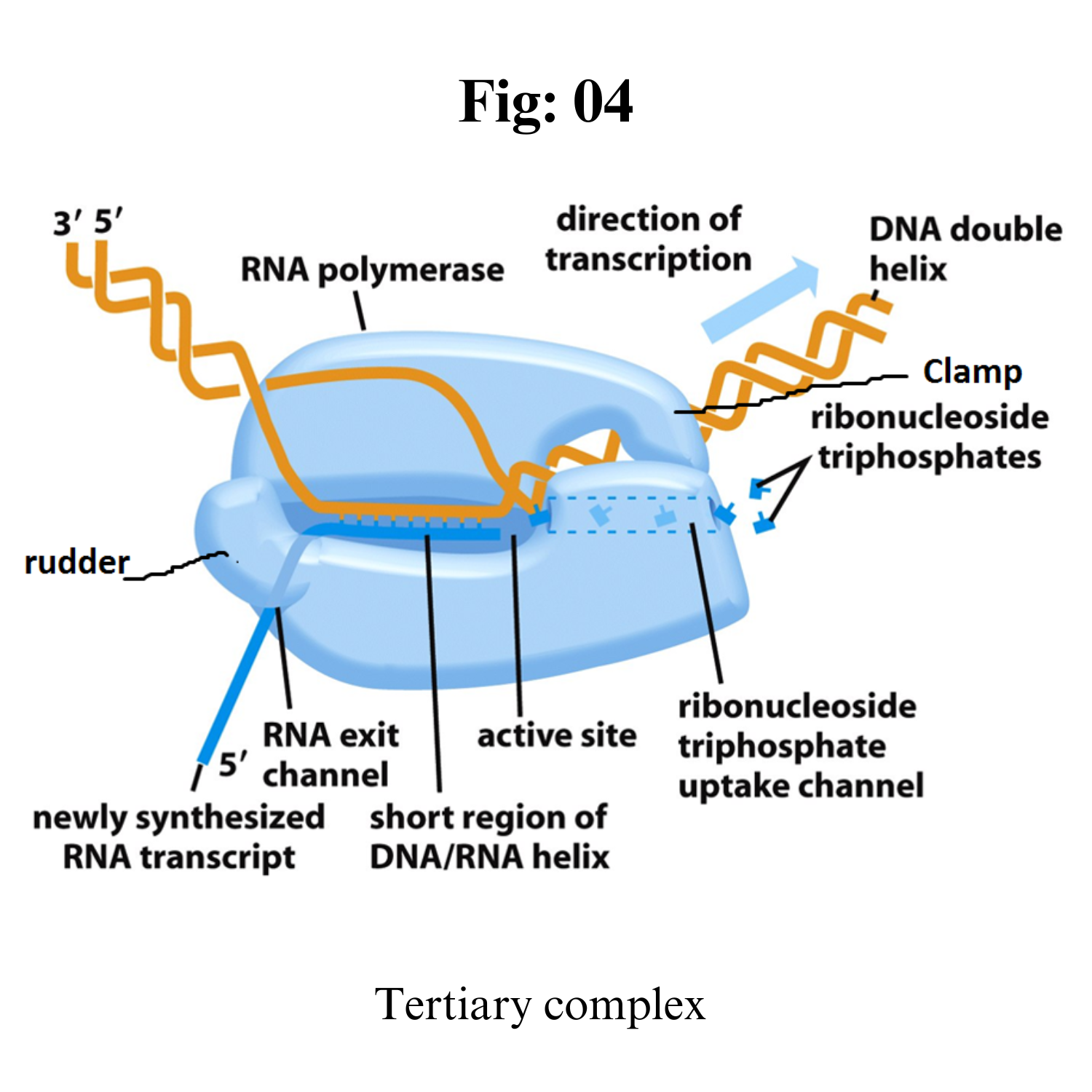
If the enzyme synthesize short RNA molecules of less than 10 bp, it does not further elongates which is called abortive initiation. This is because σ 3.2 acting as mimic of RNA and it lies at middle of RNA exit channel in open complex.
When the RNA polymerase manage to synthesize RNA more than 10 bp long, it eject the σ 3.2 region and RNA further elongates and exit from RNA exit channel. This is the formation of tertiary complex.
2. Elongation:
After synthesis of RNA more than 10 bp long, the σ-factor is ejected and the enzyme move along 5’-3’ direction continuously synthesizing RNA.
The synthesized RNA exit from RNA exit channel.
The synthesized RNA is proof reads by Hydrolytic editing. For this the polymerase back track by one or more nucleotide and cleave the RNA removing the error and synthesize the correct one. The Gre factor enhance this proof reading process.
Pyrophospholytic editing another mechanism of removing altered nucleotide.
3. Termination:
There are two mechanism of termination.
i. Rho independent:
In this mechanism, transcription is terminated due to specific sequence in terminator DNA.
The terminator DNA contains invert repeat which cause complimentary pairing as transcript RNA form hair pin structure.
This invert repeat is followed by larger number of TTTTTTTT(~8 bp) on template DNA. The uracil appear in RNA. The load of hair pin structure is not tolerated by A=U base pair so the RNA get separated from RNA-DNA heteroduplex.
ii. Rho dependent:
In this mechanism, transcription is terminated by rho (ρ) protein.
It is ring shaped single strand binding ATpase protein.
The rho protein bind the single stranded RNA as it exit from polymerase enzyme complex and hydrolyse the RNA from enzyme complex.
The rho protein does not bind to those RNA whose protein is being translated. Rather it bind to RNA after translation.
In bacteria transcription and translation occur simultaneously so the rho protein bind the RNA after translation has completed but transcription is still ON.

Prokaryotic post-transcriptional regulators typically modulate RNA decay, translation initiation efficiency or transcript elongation. Different types of prokaryotic post-transcriptional regulators have been identified, including small RNAs (sRNAs) and RNA-binding proteins (RBPs). sRNAs are typically defined as non-coding RNA molecules that bind with limited complementarity near the ribosome binding site (RBS) of their target mRNA, causing competition with the ribosome for binding to this region.
However, the number of sRNAs that deviate from this general definition is increasing. The new insights into the post-transcriptional mechanisms of sRNAs and their role in gene expression regulation were reviewed recently.
Here, RBPs involved in post-transcriptional regulation are discussed.
For some of these proteins, the mechanism of action and the targets are well described, as for CsrA and Hfq. Their post-transcriptional function in Escherichia coli was already reported almost 20 years ago. Lately, more insight was gained into the diverse mechanisms these two well-studied proteins use to regulate the expression of their target genes and how they regulate their own expression or activity in E. coli and in other bacteria.
Additional RBPs involved in post-transcriptional regulation have been identified only recently and not much is known about their post-transcriptional function. In this review, the general working mechanisms of RBPs are discussed first. Afterward, examples of well-known and recently identified proteins, from E. coli and from other bacteria, are described.
General Mechanisms of Regulatory Proteins that Act Post-Transcriptionally
Bacterial post-transcriptionally active regulatory proteins typically bind RNA molecules and regulate translation initiation, stability, and transcript elongation of their RNA targets, using different regulatory mechanisms.
These mechanisms include
(i) adaptation of the susceptibility of the target RNAs to RNases,
(ii) modulation of the accessibility of the RBS of mRNA targets for ribosome binding,
(iii) acting as a chaperone for the interaction of the RNA target with other effector molecules, and
(iv) modulation of transcription terminator/antiterminator structure formation, and will be described hereafter.
Prokaryotic Translation
As with mRNA synthesis, protein synthesis can be divided into three phases: initiation, elongation, and termination.

Initiation
Protein synthesis begins with the formation of an initiation complex. In E. coli, this complex involves the small 30S ribosome, the mRNA template, three initiation factors (IFs; IF-1, IF-2, and IF-3), and a special initiator tRNA, called tRNAMetf.
In E. coli mRNA, a sequence upstream of the first AUG codon, called the Shine-Dalgarno sequence (AGGAGG), interacts with the rRNA molecules that compose the ribosome.
This interaction anchors the 30S ribosomal subunit at the correct location on the mRNA template. Guanosine triphosphate (GTP), which is a purine nucleotide triphosphate, acts as an energy source during translation—both at the start of elongation and during the ribosome’s translocation. Binding of the mRNA to the 30S ribosome also requires IF-III.
The initiator tRNA then interacts with the start codon AUG (or rarely, GUG). This tRNA carries the amino acid methionine, which is formulated after its attachment to the tRNA. The formylation creates a “faux” peptide bond between the formyl carboxyl group and the amino group of the methionine. Binding of the fMet-tRNA-Metf is mediated by the initiation factor IF-2.
The fMet begins every polypeptide chain synthesized by E. coli, but it is usually removed after translation is complete. When an in-frame AUG is encountered during translation elongation, a non-formylated methionine is inserted by a regular Met-tRNAMet.
After the formation of the initiation complex, the 30S ribosomal subunit is joined by the 50S subunit to form the translation complex.
Elongation
In prokaryotes, we will review elongation from the perspective of E. coli. When the translation complex is formed, the tRNA binding region of the ribosome consists of three compartments. The A (aminoacyl) site binds incoming charged aminoacyl tRNAs.
The P (peptidyl) site binds charged tRNAs carrying amino acids that have formed peptide bonds with the growing polypeptide chain but have not yet dissociated from their corresponding tRNA. The E (exit) site releases dissociated tRNAs so that they can be recharged with free amino acids. The initiating methionyl-tRNA, however, occupies the P site at the beginning of the elongation phase of translation in both prokaryotes and eukaryotes.
During translation elongation, the mRNA template provides tRNA binding specificity. As the ribosome moves along the mRNA, each mRNA codon comes into register, and specific binding with the corresponding charged tRNA anticodon is ensured. If mRNA were not present in the elongation complex, the ribosome would bind tRNAs nonspecifically and randomly.
Elongation proceeds with charged tRNAs sequentially entering and leaving the ribosome as each new amino acid is added to the polypeptide chain. Movement of a tRNA from A to P to E site is induced by conformational changes that advance the ribosome by three bases in the 3′ direction. The energy for each step along the ribosome is donated by elongation factors that hydrolyze GTP.
GTP energy is required both for the binding of a new aminoacyl-tRNA to the A site and for its translocation to the P site after formation of the peptide bond. Peptide bonds form between the amino group of the amino acid attached to the A-site tRNA and the carboxyl group of the amino acid attached to the P-site tRNA.
The formation of each peptide bond is catalyzed by peptidyl transferase, an RNA-based enzyme that is integrated into the 50S ribosomal subunit. The energy for each peptide bond formation is derived from the high-energy bond linking each amino acid to its tRNA.
After peptide bond formation, the A-site tRNA that now holds the growing peptide chain moves to the P site, and the P-site tRNA that is now empty moves to the E site and is expelled from the ribosome. Amazingly, the E. coli translation apparatus takes only 0.05 seconds to add each amino acid, meaning that a 200-amino-acid protein can be translated in just 10 seconds.

Termination
Termination of translation occurs when a nonsense codon (UAA, UAG, or UGA) is encountered. Upon aligning with the A site, these nonsense codons are recognized by protein release factors that resemble tRNAs.
The releasing factors in both prokaryotes and eukaryotes instruct peptidyl transferase to add a water molecule to the carboxyl end of the P-site amino acid. This reaction forces the P-site amino acid to detach from its tRNA, and the newly made protein is released.
The small and large ribosomal subunits dissociate from the mRNA and from each other; they are recruited almost immediately into another translation initiation complex. After many ribosomes have completed translation, the mRNA is degraded so the nucleotides can be reused in another transcription reaction.

Post-Translational Modification
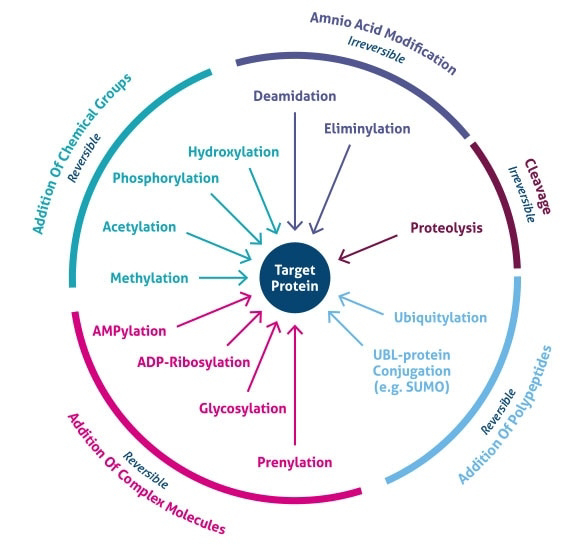
The increase in complexity from the level of the genome to the proteome is further facilitated by protein post-translational modifications (PTMs).
PTMs are chemical modifications that play a key role in functional proteomic because they regulate activity, localization, and interaction with other cellular molecules such as proteins, nucleic acids, lipids and cofactors.
PTMs estimations demonstrate that single genes encode multiple proteins. Genomic recombination, transcription initiation at alternative promoters, differential transcription termination, and alternative splicing of the transcript are mechanisms that generate different mRNA transcripts from a single gene.
Post-translational modifications are key mechanisms to increase proteomic diversity. While the genome comprises 20,000 to 25,000 genes, the proteome is estimated to encompass over 1 million proteins.
Changes at the transcriptional and mRNA levels increase the size of the transcriptome relative to the genome, and the myriad of different post-translational modifications exponentially increases the complexity of the proteome relative to both the transcriptome and genome.
Post-translational modification can occur at any step in the "life cycle" of a protein. For example, many proteins are modified shortly after translation is completed to mediate proper protein folding or stability or to direct the nascent protein to distinct cellular compartments (e.g., nucleus, membrane).
Other modifications occur after folding and localization are completed to activate or inactivate catalytic activity or to otherwise influence the biological activity of the protein. Proteins are also covalently linked to tags that target a protein for degradation.
Besides single modifications, proteins are often modified through a combination of post-translational cleavage and the addition of functional groups through a step-wise mechanism of protein maturation or activation.
Protein PTMs can also be reversible depending on the nature of the modification. For example, kinases phosphorylate proteins at specific amino acid side chains, which is a common method of catalytic activation or inactivation.
Conversely, phosphatases hydrolyze the phosphate group to remove it from the protein and reverse the biological activity. Proteolytic cleavage of peptide bonds is a thermodynamically favorable reaction and therefore permanently removes peptide sequences or regulatory domains.
Consequently, the analysis of proteins and their post-translational modifications is particularly important for the study of heart disease, cancer, neurodegenerative diseases and diabetes.
The characterization of PTMs, although challenging, provides invaluable insight into the cellular functions underlying etiological processes. Technically, the main challenges to studying post-translationally modified proteins are the development of specific detection and purification methods.
Fortunately, these technical obstacles are being overcome with a variety of new and refined proteomics technologies.

Phosphorylation
Reversible protein phosphorylation, principally on serine, threonine or tyrosine residues, is one of the most important and well-studied post-translational modifications. Phosphorylation plays critical roles in the regulation of many cellular processes, including cell cycle, growth, apoptosis and signal transduction pathways. In the following example, western blot analysis was used to evaluate phosphoprotein specificity in lysates obtained from serum-starved HeLa and NIH 3T3 cancer cell lines stimulated with epidermal growth factor (EGF) and platelet derived growth factor (PDGF), respectively.
Glycosylation
Protein glycosylation is acknowledged as one of the major post-translational modifications, with significant effects on protein folding, conformation, distribution, stability and activity. Glycosylation encompasses a diverse selection of sugar-moiety additions to proteins that ranges from simple monosaccharide modifications of nuclear transcription factors to highly complex branched polysaccharide changes of cell surface receptors. Carbohydrates in the form of asparagine-linked (N-linked) or serine/threonine-linked (O-linked) oligosaccharides are major structural components of many cell surface and secreted proteins.

Methylation
The transfer of one-carbon methyl groups to nitrogen or oxygen (N- and O-methylation, respectively) to amino acid side chains increases the hydrophobicity of the protein and can neutralize a negative amino acid charge when bound to carboxylic acids. Methylation is mediated by methyltransferases, and S-adenosylmethionine (SAM) is the primary methyl group donor.
N-acetylation
N-acetylation, or the transfer of an acetyl group to nitrogen, occurs in almost all eukaryotic proteins through both irreversible and reversible mechanisms. N-terminal acetylation requires the cleavage of the N-terminal methionine by methionine aminopeptidase (MAP) before replacing the amino acid with an acetyl group from acetyl-CoA by N-acetyltransferase (NAT) enzymes. This type of acetylation is co-translational, in that N-terminus is acetylated on growing polypeptide chains that are still attached to the ribosome. While 80 to 90% of eukaryotic proteins are acetylated in this manner, the exact biological significance is still unclear.
Acetylation at the ε-NH2 of lysine (termed lysine acetylation) on histone N-termini is a common method of regulating gene transcription. Histone acetylation is a reversible event that reduces chromosomal condensation to promote transcription, and the acetylation of these lysine residues is regulated by transcription factors that contain histone acetyltransferase (HAT) activity. While transcription factors with HAT activity act as transcription co-activators, histone deacetylase (HDAC) enzymes are co-repressors that reverse the effects of acetylation by reducing the level of lysine acetylation and increasing chromosomal condensation.
Sirtuins (silent information regulator) are a group of NAD-dependent deacetylases that target histones. As their name implies, they maintain gene silencing by hypoacetylating histones and have been reported to aid in maintaining genomic stability.
While acetylation was first detected in histones, cytoplasmic proteins have been reported to also be acetylated, and therefore acetylation seems to play a greater role in cell biology than simply transcriptional regulation. Furthermore, crosstalk between acetylation and other post-translational modifications, including phosphorylation, ubiquitination and methylation, can modify the biological function of the acetylated protein.
Protein acetylation can be detected by chromatin immunoprecipitation (ChIP) using acetyllysine-specific antibodies or by mass spectrometry, where an increase in histone by 42 mass units represents a single acetylation.




