Introduction
Perhaps the most amazing thing about mitosis is its precision, a feature that has intrigued biologists since Walther Flemming first described chromosomes in the late 1800s. Although Flemming was able to correctly deduce the sequence of events in mitosis, this sequence could not be experimentally verified for several decades, until advances in light microscopy made it possible to observe chromosome movements in living cells. Researchers now know that mitosis is a highly regulated process involving hundreds of different cellular proteins. The dynamic nature of mitosis is best appreciated when this process is viewed in living cells.
Mitosis Occupies a Portion of the Cell Cycle
In his pioneering studies of mitosis, Flemming noted that the nuclear material, which he named "chromatin" for its ability to take up stains, did not have the same appearance in all cells. (We still use the word "chromatin" today, albeit in a more biochemical sense to refer to complexes of nuclear DNA and protein.)
Specifically, in some cells, chromatin appeared as an amorphous network, although in other cells, it appeared as threadlike bodies that Flemming named "mitosen." Based on his observations, Flemming had the insight to propose that chromatin could undergo reversible transformations in cells.
Today, scientists know that Flemming had successfully distinguished chromosomes in the interphase portion of the cell cycle from chromosomes undergoing mitosis, or the portion of the cell cycle during which the nucleus divides. With very few exceptions, mitosis occupies a much smaller fraction of the cell cycle than interphase.
The difference in DNA compaction between interphase and mitosis is dramatic. A precise estimate of the difference is not possible, but during interphase, chromatin may be hundreds or even thousands of times less condensed than it is during mitosis.
For this reason, the enzyme complexes that copy DNA have the greatest access to chromosomal DNA during interphase, at which time the vast majority of gene transcription occurs.
In addition, chromosomal DNA is duplicated during a sub portion of interphase known as the S, or synthesis, phase. As the two daughter DNA strands are produced from the chromosomal DNA during S phase, these daughter strands recruit additional histones and other proteins to form the structures known as sister chromatids.
The sister chromatids, in turn, become "glued" together by a protein complex named cohesin. Cohesin is a member of the SMC, or structural maintenance of chromosomes, a family of proteins. SMC proteins are DNA-binding proteins that affect chromosome architectures; indeed, cells that lack SMC proteins show a variety of defects in chromosome stability or chromosome behavior.
Current data suggest that cohesin complexes may literally form circles that encompass the two sister chromatids. At the end of S phase, cells are able to sense whether their DNA has been successfully copied, using a complicated set of checkpoint controls that are still not fully understood. For the most part, only cells that have successfully copied their DNA will proceed into mitosis.

Chromatin Is Extensively Condensed as Cells Enter Mitosis
The most obvious difference between interphase and mitosis involves the appearance of a cell's chromosomes. During interphase, individual chromosomes are not visible, and the chromatin appears diffuse and unorganized. Recent research suggests, however, that this is an oversimplification and that chromosomes may actually occupy specific territories within the nucleus.
In any case, as mitosis begins, a remarkable condensation process takes place, mediated in part by another member of the SMC family, condensin.
Like cohesin, condensin is an elongated complex of several proteins that binds and encircles DNA. In contrast to cohesin, which binds two sister chromatids together, condensin is thought to bind a single chromatid at multiple spots, twisting the chromatin into a variety of coils and loops.

The Mitotic Spindle Aids in Chromosome Separation
During mitosis, chromosomes become attached to the structure known as the mitotic spindle. In the late 1800s, Theodor Boveri created the earliest detailed drawings of the spindle based on his observations of cell division in early Ascaris embryos.
Boveri's drawings, which are amazingly accurate, show chromosomes attached to a bipolar network of fibers. Boveri observed that the spindle fibers radiate from structures at each pole that we now recognize as centrosomes, and he also noted that each centrosome contains two small, rodlike bodies, which are now known as centrioles.
Boveri observed that the centrioles duplicate before the chromosomes become visible and that the two pairs of centrioles move to separate poles before the spindle assembles. We now know that centrioles duplicate during S phase, although many details of this duplication process are still under investigation.
The composition of the spindle fibers remained unknown until the 1960s, when tubulin was discovered and techniques were developed for visualizing spindles using electron microscopes.
It is now well-established that spindles are bipolar arrays of microtubules composed of tubulin and that the centrosomes nucleate the growth of the spindle microtubules. During mitosis, many of the spindle fibers attach to chromosomes at their kinetochores, which are specialized structures in the most constricted regions of the chromosomes.
The length of these kinetochore-attached microtubules then decreases during mitosis, pulling sister chromatids to opposite poles of the spindle. Other spindle fibers do not attach to chromosomes, but instead form a scaffold that provides mechanical force to separate the daughter nuclei at the end of mitosis.

Mitosis Is Divided into Well-Defined Phases
From his many detailed drawings of mitosen, Walther Flemming correctly deduced, but could not prove, the sequence of chromosome movements during mitosis.
Flemming divided mitosis into two broad parts: a progressive phase, during which the chromosomes condensed and aligned at the center of the spindle, and a regressive phase, during which the sister chromatids separated.
Our modern understanding of mitosis has benefited from advances in light microscopy that have allowed investigators to follow the process of mitosis in living cells. Such live cell imaging not only confirms Flemming's observations, but it also reveals an extremely dynamic process that can only be partially appreciated in still images.
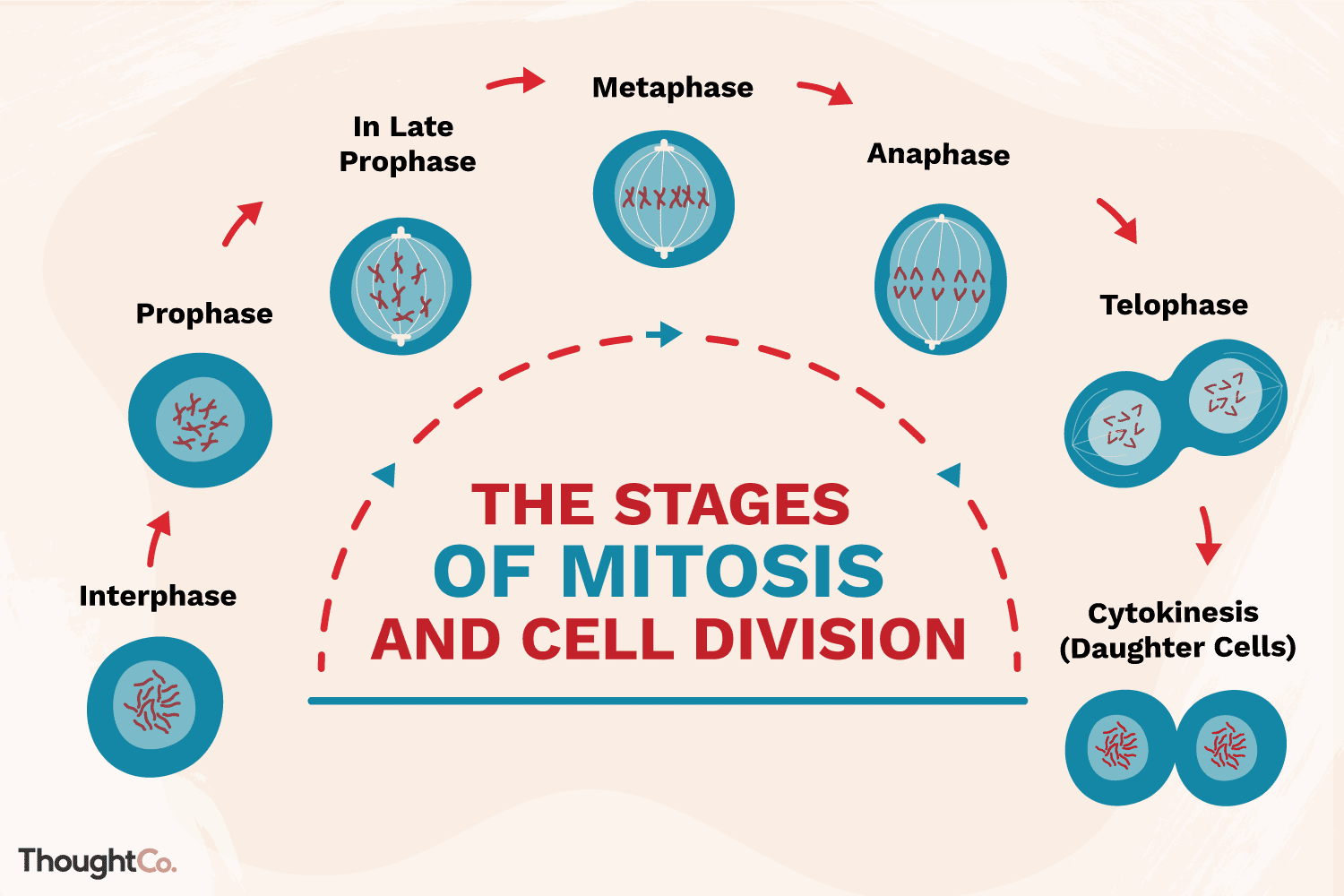
Today, mitosis is understood to involve five phases, based on the physical state of the chromosomes and spindle. These phases are prophase, prometaphase, metaphase, anaphase, and telophase.
Cytokinesis is the final physical cell division that follows telophase, and is therefore sometimes considered a sixth phase of mitosis. All phases of mitosis, as well as the flanking periods of interphase and cytokinesis before and after.
Researchers' biochemical understanding of mitotic phases has greatly increased in recent years, revealing that this highly orchestrated process involves hundreds, if not thousands, of cellular proteins.
Prophase
Mitosis begins with prophase, during which chromosomes recruit condensin and begin to undergo a condensation process that will continue until metaphase. In most species, cohesin is largely removed from the arms of the sister chromatids during prophase, allowing the individual sister chromatids to be resolved. Cohesin is retained, however, at the most constricted part of the chromosome, the centromere. During prophase, the spindle also begins to form as the two pairs of centrioles move to opposite poles and microtubules begin to polymerize from the duplicated centrosomes.
Prometaphase
Prometaphase begins with the abrupt fragmentation of the nuclear envelope into many small vesicles that will eventually be divided between the future daughter cells. The breakdown of the nuclear membrane is an essential step for spindle assembly. Because the centrosomes are located outside the nucleus in animal cells, the microtubules of the developing spindle do not have access to the chromosomes until the nuclear membrane breaks apart.
Prometaphase is an extremely dynamic part of the cell cycle. Microtubules rapidly assemble and disassemble as they grow out of the centrosomes, seeking out attachment sites at chromosome kinetochores, which are complex platelike structures that assemble during prometaphase on one face of each sister chromatid at its centromere. As prometaphase ensues, chromosomes are pulled and tugged in opposite directions by microtubules growing out from both poles of the spindle, until the pole-directed forces are finally balanced. Sister chromatids do not break apart during this tug-of-war because they are firmly attached to each other by the cohesin remaining at their centromeres. At the end of prometaphase, chromosomes have a bi-orientation, meaning that the kinetochores on sister chromatids are connected by microtubules to opposite poles of the spindle.
Metaphase
Next, chromosomes assume their most compacted state during metaphase, when the centromeres of all the cell's chromosomes line up at the equator of the spindle. Metaphase is particularly useful in cytogenetics, because chromosomes can be most easily visualized at this stage. Furthermore, cells can be experimentally arrested at metaphase with mitotic poisons such as colchicine. Video microscopy shows that chromosomes temporarily stop moving during metaphase. A complex checkpoint mechanism determines whether the spindle is properly assembled, and for the most part, only cells with correctly assembled spindles enter anaphase.
Anaphase
The progression of cells from metaphase into anaphase is marked by the abrupt separation of sister chromatids. A major reason for chromatid separation is the precipitous degradation of the cohesin molecules joining the sister chromatids by the protease separase.
Two separate classes of movements occur during anaphase. During the first part of anaphase, the kinetochore microtubules shorten, and the chromosomes move toward the spindle poles. During the second part of anaphase, the spindle poles separate as the non-kinetochore microtubules move past each other. These latter movements are currently thought to be catalyzed by motor proteins that connect microtubules with opposite polarity and then "walk" toward the end of the microtubules.
Telophase
Mitosis ends with telophase, or the stage at which the chromosomes reach the poles. The nuclear membrane then reforms, and the chromosomes begin to decondense into their interphase conformations. Telophase is followed by cytokinesis, or the division of the cytoplasm into two daughter cells. The daughter cells that result from this process have identical genetic compositions.
Cytokinesis
Cytokinesis is the physical process of cell division, which divides the cytoplasm of a parental cell into two daughter cells. It occurs concurrently with two types of nuclear division called mitosis and meiosis, which occur in animal cells. Mitosis and each of the two meiotic divisions result in two separate nuclei contained within a single cell.
Cytokinesis performs an essential process to separate the cell in half and ensure that one nucleus ends up in each daughter cell. Cytokinesis starts during the nuclear division phase called anaphase and continues through telophase.
A ring of protein filaments called the contractile ring forms around the equator of the cell just beneath the plasma membrane. The contractile ring shrinks at the equator of the cell, pinching the plasma membrane inward, and forming what is called a cleavage furrow.
Eventually, the contractile ring shrinks to the point that there are two separate cells each bound by its own plasma membrane.


Cell Cycle Checkpoints
Cell cycle checkpoints are surveillance mechanisms that monitor the order, integrity, and fidelity of the major events of the cell cycle. These include growth to the appropriate cell size, the replication and integrity of the chromosomes, and their accurate segregation at mitosis.
Many of these mechanisms are ancient in origin and highly conserved, and hence have been heavily informed by studies in simple organisms such as the yeasts.
Others have evolved in higher organisms, and control alternative cell fates with significant impact on tumor suppression. Here, we consider these different checkpoint pathways and the consequences of their dysfunction on cell fate.
The central machines that drive cell cycle progression are the cyclin-dependent kinases (CDKs). These are serine/threonine protein kinases that phosphorylate key substrates to promote DNA synthesis and mitotic progression.
The catalytic subunits are in molar excess, but lack activity until bound by their cognate cyclin subunits, which are tightly regulated at both the levels of synthesis and ubiquitin-dependent proteolysis.
Cyclin-binding allows inactive CDKs to adopt an active configuration akin to monomeric and active kinases. Layered on top of this regulation, CDK activity can also be negatively regulated by the binding of small inhibitory proteins, the CKIs, or by inhibitory tyrosine phosphorylation which blocks phosphate transfer to substrates.

The Mitotic Spindle Checkpoint
The segregation of sister chromatids at anaphase is under the mechanical control of the mitotic spindle. The spindle is composed of microtubules and several motor proteins at both the centrosomal and kinetochore ends, plus additional motors that provide force between overlapping microtubules that do not attach to kinetochores.
It is essential that spindle attachment occurs in a bi-oriented fashion such that sister chromatids are under tension at metaphase, and attached to both poles of the spindle. Once all kinetochores are attached and aligned at the metaphase plate, anaphase can proceed as is promoted by the activity of a large E3 ubiquitin ligase known as the Anaphase-Promoting Complex or Cyclosome (APC/C).
This ligase targets a number of proteins, but most essential are the mitotic cyclins, which abolishes CDK activity, and securin, the degradation of which allows separase to be released and cleave cohesin complexes at the kinetochores.
APC activity is controlled by two accessory proteins: Cdc20, which functions up to the metaphase–anaphase, and Cdh1, which continues to facilitate APC-mediated ubiquitination once cyclin and separase degradation has begun. Once sister chromatid cohesion is released, spindle tension and the associated motor proteins enable sister chromatids to move apart and form identical daughter nuclei.
The spindle checkpoint functions to prevent activation of APC Cdc20 under conditions where kinetochores are not occupied by spindle microtubules, or are attached but not under tension (for example, when attached to the same pole, known as a merotelic attachment).
Under these conditions, the spindle checkpoint protein Mad2 (Mitotic Arrest Deficient) inhibits Cdc20 activity both in the context of Cdc20 at unattached kinetochores, where it forms a mitotic checkpoint complex, and at APC-bound molecules.
Cdc20 is also regulated by the mitotic checkpoint kinase Bub1 in yeast (Budding uninhibited by benomyl) and its cousin Bub1R in mammals. As Cdc2 is inactive, so is APC, and hence, cells cannot enter anaphase.
The spindle checkpoint includes a number of other proteins, with the list growing with evolutionary complexity. In addition, the formation of the spindle and the detection and correction of spindle defects are under the control of the Polo, Aurora, and NIMA-related (Nek) kinases.
In this regard, the spindle checkpoint shares the same basic premise as those controlling DNA integrity discussed above—prevent a cell cycle transition while other effectors correct a genome-altering defect. However, the mitotic checkpoint is unique in that it functions to maintain CDK activity, whereas those functioning in interphase aim to maintain CDK inactivity.
Cell Size Control
In order to maintain cell size and ensure that each daughter cell is endowed with the appropriate amount of genetic and biosynthetic material, cells must, on average, exactly double their contents before division.
Control of cell size is critical for regulating nutrient distribution for the cell and for regulating organ size and function in multicellular organisms.
The existence of cell size checkpoints has been proposed for allowing cells to coordinate cell size with cell cycle progression. Cell size checkpoints have been observed in G1 and G2.
Early evidence for these checkpoints came from observations that the size of new daughter cells after mitosis affects cell cycle progression: large daughter cells speed up progression through G1 and/or G2, and small daughter cells delay exit from these growth phases.
However, different species and cell types vary widely in the location of these checkpoints within the cell cycle, and thus in how the cell cycle is affected in response to change in cell size.
Not surprisingly, much of what is known about size checkpoints at the molecular level is based on regulation of the proteins involved in G1 and G2/M progression.
Control of the G1 cell size checkpoint has been studied most extensively in budding yeast, where the cyclin Cln3, which activates Start, regulates cell size.
Control of the G2/M cell size checkpoint has been studied most extensively in fission yeast, where Cdc25 and Wee1 respond to cell size and nutritional status in their control of the Cdc2-cyclin B complex.
One proposed mechanism for control of cell size is via the monitoring of protein translation. Ribosomal mass, and thus translational activity, should correlate with the size of the cell, so it is thought that there is some product of translation called a “translational sizer” that increases in abundance with cell size and that exerts control over the cell cycle after a certain amount has accumulated.
Cln3 and Cdc25 are both proposed translational sizers. This hypothesis also offers an explanation for how cell size and the cell cycle respond to nutritional status.
In yeast, several signaling pathways, including the PKA and TOR pathways, are proposed to mediate nutrient control of the cell cycle, and the unifying characteristic of these pathways is that they control ribosome biogenesis, such that translational activity serves as a cellular indicator of nutritional status.
DNA Damage Responses
Throughout interphase, DNA damage elicits a cell cycle arrest that allows time for repair pathways to operate prior to commitment to subsequent phases of the cell cycle.
The source of DNA damage may be intrinsic, such as intermediates of metabolism, attrition of telomeres, oncogene overexpression, and DNA replication errors.
Alternatively, there are many extrinsic sources of DNA damage ranging from sunlight, to carcinogens, ionizing radiation or other anticancer therapeutics. While there are many lesion-specific responses for DNA repair, different lesions in genomic DNA activate common checkpoint pathways whose goal is to maintain CDKs in an inactive state until the lesion is removed.
Broadly speaking, DNA damage checkpoints can be separated into those controlled by the tumor suppressor and transcription factor p53, and those ultimately under the control of the checkpoint kinase Chk1, and we will consider the latter first.
In higher organisms, the transcription factor p53 is a critical component of DNA damage checkpoints, particularly in G1 phase. p53 is regulated by a plethora of posttranslational modifications, including N-terminal phosphorylation on serine-15, which is catalyzed by ATR and its cousins ATM (Ataxia Telangiectasia Mutated) and DNA-PKcs (DNA-dependent protein kinase, catalytic subunit).
Similar to ATR, these kinases are targeted to double-strand DNA breaks by interacting proteins: the MRN (Mre11-Rad50-Nbs1) complex for ATM, and the Ku70–Ku80 complex for DNA-PKcs.
Activated p53 is stabilized through protection from its E3 ubiquitin ligase Mdm2, and as a tetramer transactivates the expression of a large number of genes, including the cyclin-dependent kinase inhibitor (CKI) p21.
Through this mechanism, G1 CDKs are inhibited, and DNA damage is repaired prior to DNA replication. However, p53 can also repress the expression of genes, and is required for prolonged G2 arrest in the face of persistent DNA damage.
Moreover, p53 can direct the alternative cell fates of apoptosis or senescence. Indeed, the cell cycle arrest function of p53 seems to be a later adopted function, as Drosophila p53 regulates apoptosis, but not cell cycle progression.
Monitoring DNA Replication
S phase marks a particularly vulnerable time for cells to cope with DNA damage. Not only must lesions be repaired as in G1 and G2 cells, but they also act as a physical impediment to the replicative polymerases.
DNA replication is initiated at specific sites, the replication origins. These are epigenetically defined by a number of proteins that ensure they fire (start replicating) once and only once per cell cycle.
Replication origin firing is controlled by the phosphorylation of two proteins, Cdt1 and Cdc6, which is catalyzed by both CDKs and the Dbf4-dependent protein kinase (DDK) Cdc7. Such phosphorylation not only initiates replication but also leads to degradation of these proteins, and hence the origin cannot be refire.
When the polymerase and its associated proteins (the replisome) encounter a blockade to progression, it is imperative that the replisome remains stably associated with the replicating chromatid so that replication can resume once the blockade is removed. Such blockades can be modified dNTPs, abasic sites, protein–DNA complexes, or result from the depletion of dNTPs. This replisome stabilization is the function of the intra-S-phase checkpoint.
The effector kinase of the intra-S-phase checkpoint is known as Cds1 in fission yeast or Chk2 in humans. Despite its related name, Chk2 is not biochemically or functionally related to Chk1.
Cds1/Chk2 has an N-terminal phospho-S/T-binding Forkhead-Associated (FHA) domain followed by a kinase domain. Upon replication stalling, the replisome component Mrc1 (Mediator of the replication checkpoint) is phosphorylated by ATR.
This creates a binding site for Cds1/Chk2, which is then phosphorylated by ATR, and then fully activated by autophosphorylation. Activated Cds1/Chk2 then stabilizes the stalled replisome by phosphorylation of several subunits, notably the MCM helicase.
In budding yeast, the Rad53 kinase serves the function of Cds1/Chk2. Like Cds1/Chk2, Rad53 has an N-terminal FHA domain followed by a kinase domain. However, Rad53 has an additional C-terminal FHA domain not seen in Cds1 that is important in its activation by DNA damage.
Summary
Mitosis is a part of cell division during which the nucleus of a cell is divided into two daughter nuclei having an identical number of chromosomes.
The cell cycle between the two subsequent cell divisions is called interphase, and consists of the following phase;
G1 phase, during which a cell grows and decides whether to divide or not
S phase, during which the DNA of a cell is replicated resulting in duplication of chromosomes
G2 phase, during which the proteins necessary for mitosis are formed by the cell
The cells that do not receive a signal for cell division enter the quiescent G0 phase.
The process of mitosis consists of the following phases;
Prophase, during which the nuclear envelope degenerates, chromosomes become prominent, and the mitotic apparatus starts forming.
Metaphase: In this phase, mitotic spindles are formed, attaching to the chromatids, pulling them and aligning along the mitotic plate.
Anaphase; During this phase, chromosomes are segregated, and sister chromatids are pulled towards the opposite poles. Cells also begin to elongate.
Telophase, it results in the formation of the nuclear envelope around the sister chromatids and two nuclei are formed
Mitosis is followed by cytokinesis that divides the cell into two daughter cells.
Animal cell is divided by cleavage furrow
Plant cell is divided by formation of fusion plate via Golgi-derived vesicles
The process of mitosis have the following significance in the life of organisms;
It is involved in both intrauterine and extrauterine growth and development
It is involved in the replacement of the cells that are continuously sloughed off
The process of regeneration is dependent on mitosis
Asexual reproduction takes place via mitosis.



